


The call came at 2 AM. Marcus, CEO of a promising cardiac device startup, was calling from his car outside the FDA building in Maryland. His voice was shaking.
“They rejected our 510(k),” he said. “After 18 months and $2.3 million, they want us to run clinical trials. Our investors are pulling out. We’re done.”
Marcus had made the same fatal mistake I see repeatedly: he budgeted for regulatory approval like it was a predictable engineering problem. Build device, submit paperwork, get approval. In reality, the FDA regulatory process is more like navigating a maze blindfolded, where each wrong turn costs hundreds of thousands of dollars and months of delay.
The regulatory nightmare isn’t just about the complexity—it’s about the exponential cost escalation that catches entrepreneurs completely off guard. What seems like “just more documentation” for Class II and III devices often costs more than the entire product development phase.
The Regulatory Classification Reality Check
Most medical device entrepreneurs understand there are three device classes, but they dramatically underestimate the cost and complexity differences between them. It’s not a linear progression—it’s exponential.
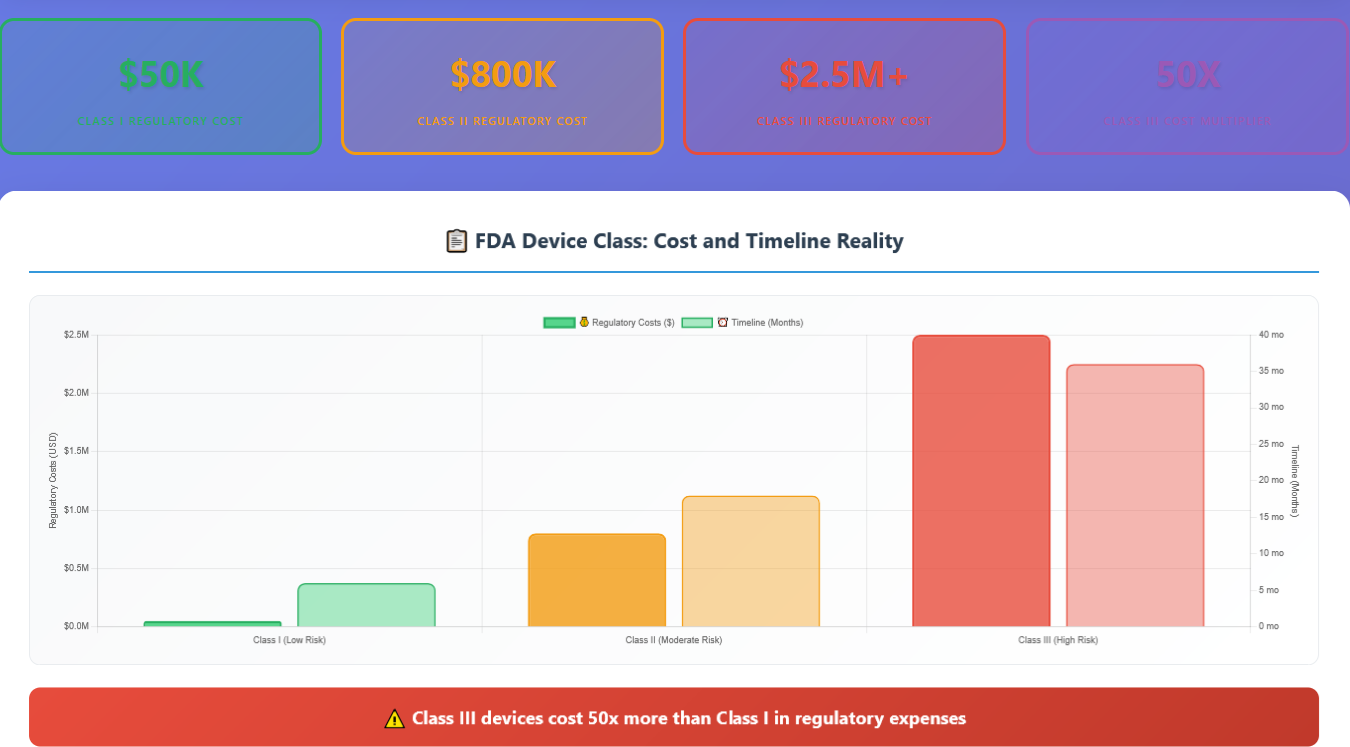
Class I Devices ($50K regulatory cost, 6 months): Most are exempt from premarket notification. Simple documentation, basic quality systems, minimal clinical evidence required.
Class II Devices ($800K regulatory cost, 18 months): Require 510(k) premarket notification, predicate device comparison, clinical data, comprehensive quality systems. This is where most entrepreneurs get blindsided.
Class III Devices ($2.5M+ regulatory cost, 36+ months): Require Premarket Approval (PMA), extensive clinical trials, comprehensive safety and effectiveness data, ongoing post-market surveillance.
The jump from Class I to Class II isn’t just 16x more expensive—it requires completely different expertise, processes, and strategic thinking.
The 510(k) Money Pit
The 510(k) process is where most Class II device companies discover that “substantially equivalent” doesn’t mean “easy approval.” The FDA received over 4,000 510(k) submissions in 2023, and the process has become increasingly complex and expensive.
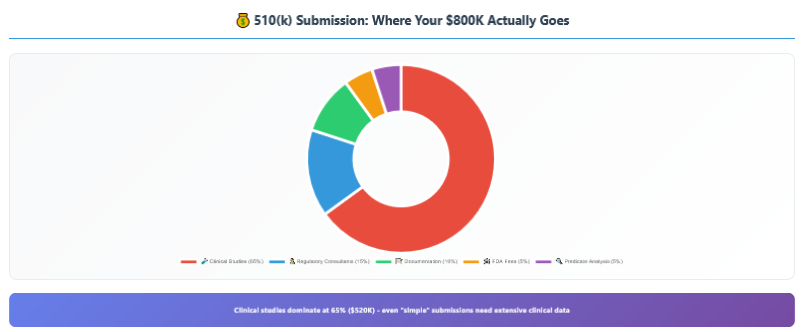
Clinical Studies (65% – $520K average): Even “simple” 510(k)s often require clinical data. Biocompatibility testing, usability studies, performance testing, and comparative effectiveness studies add up quickly.
Regulatory Consultants (15% – $120K average): Experienced regulatory professionals charge $300-500/hour. Quality regulatory guidance is expensive but essential for first-time submissions.
Documentation & Writing (10% – $80K average): The 510(k) document itself is a complex technical writing project requiring specialized skills and extensive formatting.
FDA Fees & Administrative (5% – $40K average): FDA user fees, quality system audits, and administrative overhead.
Predicate Analysis (5% – $40K average): Identifying and analyzing predicate devices, competitive intelligence, and regulatory landscape analysis.
Total typical 510(k) cost: $800K
And this assumes your submission is approved on the first try. FDA rejection rates vary by device type, but additional information requests and resubmissions can double these costs.
The Clinical Trial Black Hole
For many Class II devices and all Class III devices, clinical trials represent the biggest regulatory expense—and the most unpredictable. Clinical trials aren’t just expensive; they’re exponentially expensive if anything goes wrong.
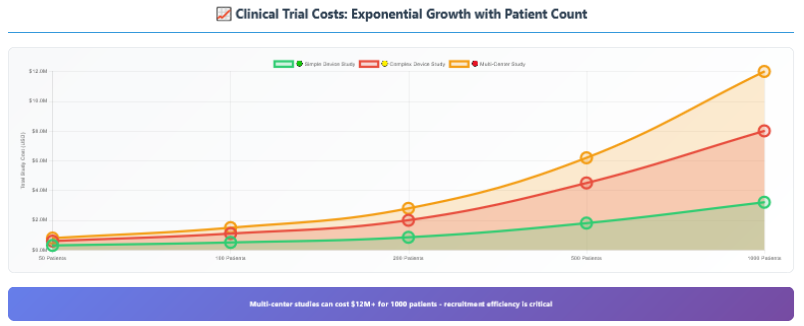
The cost components that drive clinical trial expenses:
Site Fees: Clinical research sites charge $15K-50K per site for setup, plus $2K-10K per patient enrolled. Multi-center studies multiply these costs across multiple locations.
Principal Investigator Fees: Experienced clinicians charge $500-2000 per patient for their time and expertise.
Patient Recruitment: Finding and enrolling patients costs $1K-5K per patient, depending on the indication and inclusion criteria.
Monitoring and Quality Assurance: Clinical research organizations (CROs) charge $200-400/hour for study monitoring, data management, and quality assurance.
Regulatory Oversight: Institutional Review Board (IRB) fees, FDA communications, and regulatory compliance monitoring.
Statistical Analysis: Final study analysis, report writing, and regulatory submission preparation.
The Timeline Nightmare
The financial costs are just one part of the regulatory nightmare. The timeline uncertainty can kill companies that run out of cash waiting for approval.
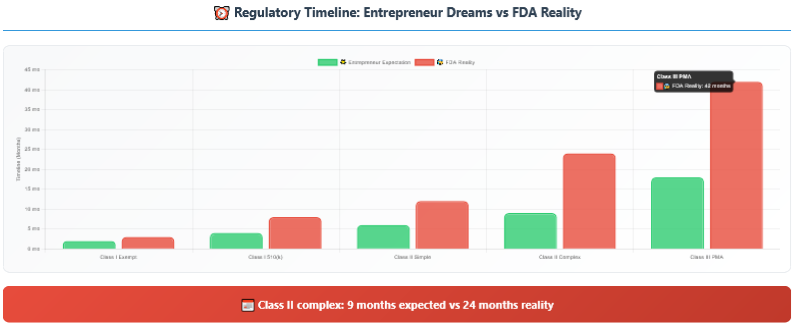
Why timelines are so unpredictable:
FDA Review Variability: The FDA’s official 510(k)
review time is 90 days, but that doesn’t include time for additional
information requests, which occur in 60-80% of submissions.
Clinical Trial Duration: Patient recruitment often
takes 2-3x longer than expected. Protocol amendments, adverse events, and site
management issues add months.
Regulatory Changes: FDA guidance evolves constantly.
Requirements that didn’t exist when you started may be mandatory by the time
you submit.
Quality System Issues: FDA inspections can reveal
quality system deficiencies that require months to address before approval.
The International Regulatory Maze
Many entrepreneurs think they can offset FDA costs by
pursuing international approvals first. This strategy often backfires
spectacularly.

European Union (MDR): The Medical Device Regulation implemented in 2021 has made European approval more expensive and complex than FDA in many cases. Post-market surveillance requirements are particularly burdensome.
Canada (Health Canada): Often seen as an “easier” first approval, but clinical data requirements have increased significantly, and the Canadian market alone rarely justifies the investment.
Multiple Jurisdictions: Each regulatory body has different requirements. Clinical data accepted by one may not be sufficient for another, leading to duplicated studies and costs.
The Hidden Regulatory Costs
Beyond the obvious expenses of submissions and clinical trials, regulatory compliance creates dozens of hidden costs that compound over time.

Quality System Maintenance ($120K annually): ISO 13485 compliance, document control, training, internal audits, and management review processes require dedicated resources.
Post-Market Surveillance ($80K annually): Monitoring device performance, collecting clinical data, trending adverse events, and maintaining clinical evidence.
Design Change Controls ($60K annually): Every design change requires formal evaluation, documentation, and often regulatory notification or approval.
Regulatory Updates ($40K annually): Staying current with evolving regulations, guidance documents, and industry standards requires dedicated regulatory expertise.
Inspection Readiness ($30K annually): Maintaining FDA inspection readiness, mock audits, and corrective action response capabilities.
Adverse Event Reporting ($25K annually): Systems for collecting, investigating, and reporting adverse events and device malfunctions.
Real-World Regulatory Disasters
Let me share three regulatory disasters I’ve witnessed that illustrate how badly things can go wrong:
Case 1: The $3M 510(k) Nightmare A orthopedic device company submitted what they thought was a straightforward 510(k). FDA requested additional clinical data. The clinical study revealed safety issues requiring design changes. The design changes required new predicate analysis. Total cost: $3.2M over 4 years. The company eventually sold their technology to a larger player for less than they spent on regulatory approval.
Case 2: The PMA Pivot A Class II cardiac device received an FDA letter stating their device should be regulated as Class III, requiring PMA instead of 510(k). This changed their regulatory timeline from 18 months to 4+ years and their budget from $800K to $5M+. Investors pulled funding, and the company folded.
Case 3: The International Nightmare A company pursued CE marking first, spending $600K and 18 months for European approval. When they submitted to FDA, they were told none of their clinical data was acceptable for US approval due to different study design requirements. They needed to run completely separate US clinical trials, essentially doubling their regulatory costs.
Survival Strategies for Regulatory Reality
The regulatory nightmare is real, but it’s survivable with the right strategy and realistic expectations.
- Early FDA Engagement
Pre-Submission Meetings: FDA Q-Sub meetings cost $25K but can save hundreds of thousands by clarifying requirements upfront. Use them liberally.
Regulatory Strategy Development: Invest in experienced regulatory consultants early in development, not after your device is designed. Regulatory requirements should drive design decisions, not the other way around.
- Realistic Budgeting
Triple Your Estimates: Whatever your initial regulatory budget, triple it. This accounts for the high probability of additional studies, design changes, and submission delays.
Phased Funding: Structure your funding to match regulatory milestones. Don’t assume you can raise money during regulatory review periods.
- Quality System Foundation
Start Early: Implement ISO 13485 quality systems during development, not after. It’s much cheaper to build compliance in than to retrofit it.
Design Controls: Follow FDA design control requirements from day one. This prevents costly rework and creates the documentation foundation for your submissions.
- Clinical Strategy
Minimize Risk: Design your clinical studies to minimize the chance of negative results. Use experienced clinical investigators and conservative study designs.
Statistical Power: Ensure your studies are properly powered for regulatory success, not just statistical significance. Underpowered studies are worthless for regulatory purposes.
- Strategic Partnerships
Regulatory Expertise: Partner with companies that have regulatory expertise and FDA relationships. Their guidance can prevent expensive mistakes.
Clinical Networks: Work with established clinical research networks that have experience in your device category.
The Post-Approval Reality
Getting FDA approval isn’t the end of your regulatory expenses—it’s just the beginning of a new phase of ongoing compliance costs.
Annual FDA Fees: Device registration and listing fees, quality system inspection fees, and user fees for post-market submissions.
Post-Market Studies: FDA may require post-market clinical studies as a condition of approval, adding millions in ongoing costs.
Vigilance Systems: Adverse event monitoring, trending analysis, and regulatory reporting require dedicated resources.
International Maintenance: If you’ve pursued international approvals, each jurisdiction has ongoing compliance requirements and fees.
The Bottom Line
Regulatory approval for medical devices isn’t just expensive—it’s exponentially expensive as you move from Class I to II to III. The entrepreneurs who survive understand that regulatory costs often exceed development costs and plan accordingly.
The regulatory nightmare isn’t about FDA being unreasonable—it’s about the inherent complexity of proving safety and effectiveness for devices that could harm people if they fail. The costs are high because the stakes are high.
In our final article, we’ll explore exit strategy reality—who actually buys medical device companies, what they’re looking for, and why “we’ll sell before commercialization” might be the most expensive assumption you can make.
Remember: You can’t shortcut regulatory approval, but you can plan for it realistically. Budget for the nightmare, and you might just survive it.